Abstract
Formalin fixation is a mainstay of modern histopathologic analysis, yet the practice is poorly standardized and a significant potential source of preanalytical errors. Concerns of workflow and turnaround time drive interest in developing shorter fixation protocols, but rapid protocols can lead to poor histomorphology or inadequate downstream assay results. Additionally, assays such as immunohistochemistry for phosphorylated epitopes have historically been challenging in the context of formalin-fixed tissue, indicating that there may be room for improvement in this process that is fundamental to the practice of anatomic pathology. With these issues in mind, we studied basic formalin biochemistry to develop a novel formalin fixation protocol that involves a pre-incubation in subambient temperature formalin prior to a brief exposure to heated formalin. This new protocol is more rapid than standard protocols yet preserves histomorphology and yields tissue that is compatible with an expanded set of downstream clinical and research assays, including immunohistochemistry for phosphorylated epitopes.
Introduction
In the 1890s, the industrial precursor to Schering AG patented an aqueous solution of gaseous formaldehyde with the name “formalin,” and for more than a century, histologists have used formalin to prepare tissue for sectioning and microscopic examination. Despite this long history of use, the chemistry of formaldehyde’s interactions with tissue constituents is understood less completely than one might expect. What has been thought to occur when formalin fixes tissue is that formaldehyde crosslinks proteins by creating methylene bridges between adjacent amino groups on proteins, thereby lending the tissue structural integrity as well as inactivating potentially destructive enzymes within the tissue. This is an oversimplification, however, and the interactions of formaldehyde with every type of biomolecule in a tissue are still poorly understood. Tissue proteins, for example, are traditionally thought to be simply cross-linked by formaldehyde, but they can participate in more chemical reactions with formaldehyde than just cross-linking. Protein amino groups are not the sole targets of formalin as evidenced by the observations that peptides lacking amino groups can be reversibly “fixed” and non-peptides such as nucleic acids also appear to be altered by fixation by incompletely understood mechanisms.
The behavior of formaldehyde in tissues is further muddied by the fact that formaldehyde in solution is, in fact, mostly not formaldehyde. Rather, it is in equilibrium with a large excess of its nonreactive hydrate , methylene glycol, such that only a tiny fraction of the molecules in “formalin” are reactive formaldehyde species. This latter fact may account for the oft-repeated observation that formalin penetrates tissues briskly but fixes them slowly . Despite these mechanistic uncertainties, formalin remains an overwhelmingly popular choice of fixatives in clinical laboratories , despite the existence of multiple alternative fixation strategies.
The search for ever-faster formalin fixation protocols has been driven historically by considerations of overall laboratory turnaround time and workflow, as formalin fixation and tissue processing can comprise the majority of the time required for histopathologic analysis. Commercial tissue processors designed to address this need fix tissue rapidly by applying formalin and external energy in the form of heat or microwaves . Heat likely speeds fixation for two reasons: first, the formaldehyde-methylene glycol equilibrium shifts towards formaldehyde at higher temperatures and raises the effective concentration of the active molecule and second, protein crosslinking, like many other chemical reactions, should proceed more quickly at elevated temperatures. Ultrasound has been applied to fixation, as well, as an alternate mechanism to impart energy to speed fixation .
While accelerated fixation with heated formalin has potential benefits in decreasing clinical turnaround times, our experience and that of others indicates that the quality of histologic and molecular studies suffers in heated protocols . Additionally, it is concerning that outside of a few clinical applications, tissue fixation is not rigidly standardized in the clinical laboratory. One laboratory may choose to fix clinical tissue samples rapidly at an elevated temperature and another may choose to fix slowly at ambient or low temperatures, but both preanalytical conditions are expected to perform adequately for downstream tissue-based assays. The influence of variation in preanalytical specimen preparation is increasingly being recognized as a major problem in diagnostic pathology , as it prevents accurate targeted analysis of a patient’s specimen and elucidation of the best possible therapy for a disease, both essentials in the paradigm of personalized medicine. Posttranslational protein modifications such as phosphorylation, for example, are known to be important indicators of signaling pathway activity and represent promising clinical biomarkers, but they can be very difficult to measure in fixed tissues because of preanalytical variables . Formalin fixation prior to phosphoprotein analysis must crosslink tissue proteins and inactivate the phosphatase enzymes that might remove protein phosphates; failure on either of these tasks in inappropriately fixed tissue makes it impossible to interrogate these pathways and use such assays as diagnostics.
Standardized fixation parameters have been adopted for some clinical biomarker assays to mitigate the errors stemming from preanalytical variation, such as the American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) guidelines for HER2 IHC that call for fixation in neutral buffered formalin for at least 6 hours and no more than 48 hours. While these guidelines are well intentioned, they still allow an 8-fold variation in fixation time and are not meant to represent optimal conditions for all IHC assays. It is highly unlikely, however, that the large number of different laboratory-developed IHC assays performed clinically around the world could be validated for all potential fixation protocols. Faced with this problem, we sought to identify a standardized formalin fixation protocol that was applicable to a broad range of tissue types and optimized both for speed and preservation of diagnostic molecular features. Beginning with basic protein biochemistry studies, we rationally developed a novel fixation protocol based on formalin biochemistry, demonstrated its broad applicability to standard histopathology, and finally showed that the novel protocol provided acceptable IHC assay results for numerous targets including several phosphoprotein assays.
Materials and Methods
Tissue Samples
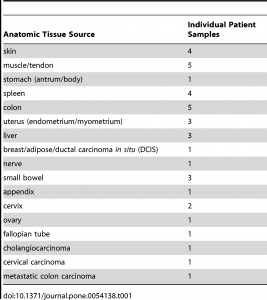
De-identified unfixed human tonsil tissue samples were obtained from either the Cooperative Human Tissue Network (CHTN, Nashville, TN) or Bio-Options Inc. (Brea, California); surgical samples were wrapped in saline-soaked gauze and kept on wet ice overnight prior to use. Human colon carcinoma samples for phosphoprotein analysis were obtained from Indivumed GmBH (Hamburg, Germany) as paraffin-embedded samples; samples from Indivumed were used because the cold ischemia time offered by this tissue vendor were superior (shorter) than possible from other vendors or in our clinical area. Indivumed scientists performed the fixation protocols described on these colon carcinoma samples. Calu-3 xenograft tumors were provided by the Assay Development Support Service (ADSS) at Ventana Medical Systems, Inc. (Tucson, AZ). Additional tissue samples were collected from surgeries at the author’s (GSB) institution under a waiver of consent, using procedures approved by the University of Washington Institutional Review Board. Upon excision, fresh tissue was carried to the pathology laboratory, generally within 30–60 minutes, and after the diagnostic pathologist had taken sections needed for diagnosis, excess tissue was dissected into several equivalently sized portions for each fixation condition. Tissue specimens were limited to 1 cm in maximum dimension, and never exceeded 4 mm in thickness. For comparison of histomorphology and IHC between tissue fixed with experimental conditions and tissue generated by pathology department histotechnologists in a CLIA- and College of American Pathologists (CAP)-certified laboratory, unstained slides from the clinical tissue block generated in each case (10–48 hours RT formalin fixation) were collected.
Fixation Protocols
For all experiments, fixation consisted of immersion in 10% neutral buffered formalin (Saturated aqueous formaldehyde from Fisher Scientific, Houston, TX, buffered to pH 6.8–7.2 with 100 mM phosphate buffer) for 0–24 hours at temperatures ranging from 4°C to 50°C. Room temperature ranged from 20–25°C. Tissue samples collected from clinical cases were fixed at room temperature for 0, 2, 4 or 24 hours, or else 2 hours in 4°C formalin followed by 2 hours in 45°C formalin, the latter protocol being termed “2+2”. The 0 hour formalin specimen was placed directly into 70% ethanol. Fixation was carried out in 100–500 mL covered beakers in a refrigerator for 4°C treatment, in a fume hood for room temperature treatment, and in a fume hood on a hot plate for elevated temperatures (i.e. 45°C). 2+2 samples were carried from the refrigerated beaker to the fume hood for sequential temperature treatments. Clinical blocks were fixed at room temperature for 10–48 hours in the clinical laboratory, with 10 hours being the minimum for weekday samples and 48 hours only used for samples fixed over weekends. To facilitate processing all research samples from a day on the same processor cycle, all samples were stored in 70% ethanol after variable-length fixation prior to starting the tissue processor (Peloris, Leica, Germany). The processor run was programed as follows: 80% ethanol for 30 minutes, 95% ethanol for 60 minutes, 100% ethanol for 3 hours, xylene for 3 hours, and paraffin for 3 hours 20 minutes. Clinical blocks were processed by the same protocol without a variable 70% ethanol preincubation. All solvent processor steps were kept at 45°C under ambient pressure, and the paraffin step was held at 60°C under vacuum. All specimens were embedded in paraffin and cut onto glass slides as 5-micron sections for routine Hematoxylin/Eosin (H&E) stain or and 4-micron sections for IHC or molecular studies. The clinical tissue samples collected for this study are enumerated in table 1.
RNase A Crosslinking
5 mg/ml RNaseA (Sigma-Aldrich, St. Louis, MO) was mixed with 10% Neutral Buffered Formalin (Richard-Allan Scientific, Kalamazoo, MI) for 10 minutes. At specified times, samples were diluted 10-fold with ice cold water. A 10 µl aliquot was then removed, mixed with an equal volume of 2× SDS-protein loading buffer, electrophoresed on a 4–15% gradient Tris-HCl-SDS polyacrylamide gel (Bio-Rad, Hercules, CA) and silver-stained according to manufacturer’s directions (EMD, Darmstadt, Germany). Precision Plus molecular weight markers were used as directed (Biorad, Hercules, CA).
Histologic Assessment
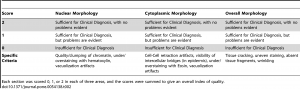
Histologic assessment of fixation quality was scored using a modification of a scoring system published by the manufacturer of the tissue processor used in this study , described intable 2. The pathologist grading the histologic appearances was blinded to the fixation protocol for each tissue section. Differences in histologic quality scores were assessed with the Kruskal-Wallis test and Dunn’s post-test to correct for multiple comparisons. For data in Figure S1, subjective histologic quality was assessed by a pathologist and categorized as “unacceptable” (colored red in figure), “acceptable for diagnosis but with recognizable defects in morphology” (colored yellow in figure), and “acceptable” (colored green in figure). To ensure reproducibility in this subjective assessment, samples fixed with different protocols were compared to control samples that had been fixed in room temperature formalin for 0, 2, 4, 8 and 24 hours. The results presented here are representative of 10 repeated experiments for each H&E assessments and 2 repeated experiments for the IHC experiments.
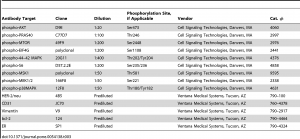
Automated Immunohistochemistry Staining
Immunohistochemistry assays were performed on a VENTANA Discovery XT automated staining instrument according to the manufacturer’s instructions. Slides were de-paraffinized using EZprep solution (Ventana Medical Systems, Inc.) at 90°C, and all reagents and incubation times were chosen as directed on antibody package inserts. Slides were developed using the OmniMap DAB detection kit (Ventana Medical Systems, Inc.) and counterstained with hematoxylin. Antibodies, clones, and titers are listed in table 3. Antibody titers were determined for each antibody using positive and negative control tissues following the manufacturer’s instructions.